

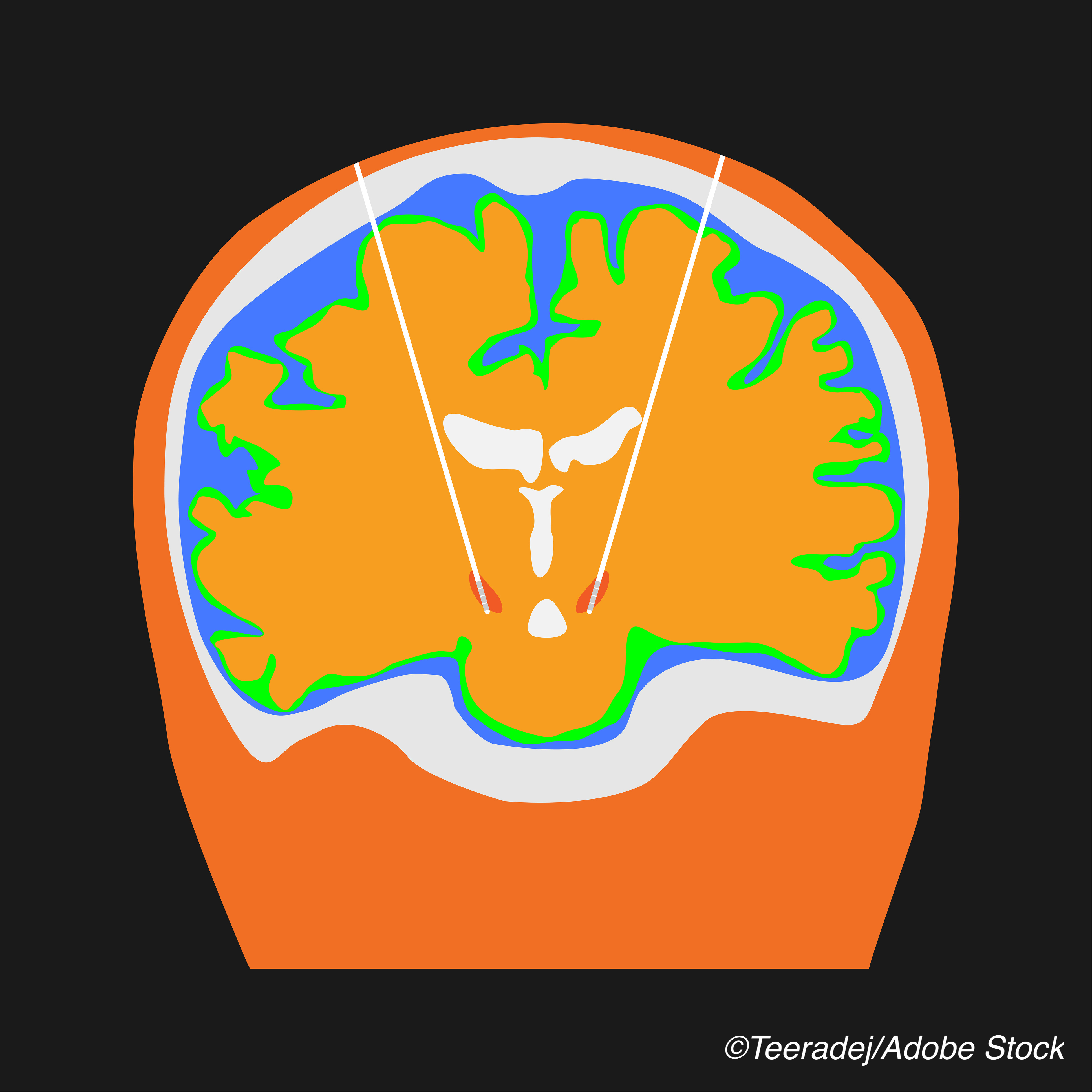
WASHINGTON — The FDA approved an updated version of a deep brain stimulation device to help treat uncontrolled symptoms in patients with moderate to advanced Parkinson’s disease (PD) that is responsive to levodopa.
The device, Boston Scientific Corporation’s fourth generation Vercise Genus Deep Brain Stimulation (DBS) System, works to treat PD symptoms by “delivering targeted electrical stimulation via surgically-implanted leads in the brain connected to [implantable pulse generators]” that power Cartesia Directional Leads, the manufacturer explained in a press release. The device is approved for conditional use in a magnetic resonance imaging environment, the company added. An earlier version of the device was first approved in the U.S. in 2017
The device is indicated for use in the bilateral stimulation of the subthalamic nucleus and the internal globus pallidus as an adjunctive therapy in reducing some of the symptoms of moderate to advanced levodopa-responsive PD that are not adequately controlled with medication. The FDA’s approval of this DBS device was based on results from the INTREPID trial, a multicenter, double-blind, randomized, sham-controlled study, the results of which were previously reported by BreakingMED. The study enrolled idiopathic Parkinson’s disease patients from May 2013-Nov. 2017 across 23 sites. Participants had over five years of motor symptoms and were 22-75 years old, with stable medication regimens from 28 days prior to consent. The trial met its primary endpoint, with patients in the active group achieving a significant mean difference in “on” time — mean daily waking hours with good symptom control, no troublesome dyskinesias, and no increase in anti-parkinsonian medications — compared to the control group at 3 months (3.03 hours from baseline, 95% CI 1.3-4.7; P<0.0001), study authors Jerrold Vitek, MD, of the University of Minnesota, and colleagues reported. In the same trial, 26 adverse events were reported in 20 patients at 3 months — 18 in the active group and eight in the control group. Adverse events included aphasia (n=1), post-operative confusion (n=4), seizure (n=2), infection (n=9), hypoventilation (n=1), implant site edema (n=2), intracranial hypotension (n=2), myocardial infarction (n=1), pneumocephalus (n=2), fever of unknown origin (n=1), and wound hemorrhage (n=1). “The site of hardware infection was cranial in all seven patients, one of whom also had evidence of infection involving the implantable pulse generator pocket,” Vitek and colleagues wrote. Serious adverse events from 3 to 12 months included implant site edema (n=2), wound infection (n=1), wound dehiscence (n=2), and mania (n=1), they added. John McKenna, Associate Editor, BreakingMED™ Cat ID: 37 Topic ID: 82,37,730,37,192,725,925Author