
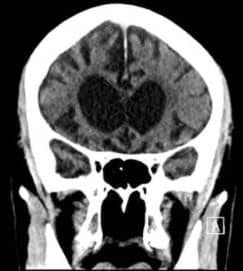
Disorders of neuronal migration during embryogenesis lead to lissencephaly (LIS), a cortical deformity defined by a smooth or virtually smooth cerebral surface and a lack of gyral and sulcal development. The PAFAH1B1 disease-linked gene product is one of several involved in neuronal migration. Lack of identifiable clinical signs makes LIS a rare disease; for example, it may manifest as early-onset epilepsy, developmental delay, or motor abnormalities similar to those seen in cerebral palsy. Since high-throughput sequencing has been shown to enhance diagnostic accuracy, researchers decided to use this method to treat this patient. They discussed the case of a 7-year-old male with non-specific clinical symptoms probably compatible with lissencephaly due to an unidentified uncommon condition. The patient participated in a study that required whole-genome sequencing. A bioinformatics workflow was used to decipher the sequence data. To prioritize the variants that were retrieved, they were first annotated. There was also research into the mitochondrial genome. The index case phenotype was explained by the discovery of a novel potential frameshift insertion in the previously characterized PAFAH1B1 gene. Based on the in silico analysis, it was found to be harmful and to trigger nonsense-mediated pathways with a high degree of certainty. A premature stop codon is generated due to the insertion, which substantially impairs protein function and likely results in the silencing of one allele. This mutation was not passed down from the healthy mother, and the unaffected paternal grandfather was not accessible for testing. This investigation led them to the discovery of a unique de novo mutation in the LIS1/PAFAH1B1 gene, the likely cause of a rare condition in a young child with non-specific clinical signs. Since the loss of function in the gene product has been described in this condition, the discovered mutation coincides with the phenotype examined. A de novo disease mechanism is hypothesized since no additional mutations in PAFAH1B1 exist at a low population frequency and because of family history.
Source: bmcpediatr.biomedcentral.com/articles/10.1186/s12887-022-03595-6