
A study evaluating the drug risdiplam demonstrated positive results in infants with type 1 spinal muscular atrophy (SMA).
Type 1 SMA is a muscle wasting disease that is the leading cause of genetic death in infants and affects one out of every 6,000 to 10,000 live births. Infants with SMA do not typically live beyond their second birthday and are unable to develop motor skills like speaking or sitting, according to Giovanni Baranello, MD, PhD, Great Ormond Street Institute of Child Health, London, U.K., and colleagues who undertook the FIREFISH study — an open-label, two-part study that assessed the efficacy of risdiplam in infants age 1-7 months with SMA.
These infants also experience respiratory infections due to neuromuscular weakness, as well as an inability to swallow. Thus, according to the authors, they will typically require feeding support, or combined feeding and ventilatory support, by 12 months of age.
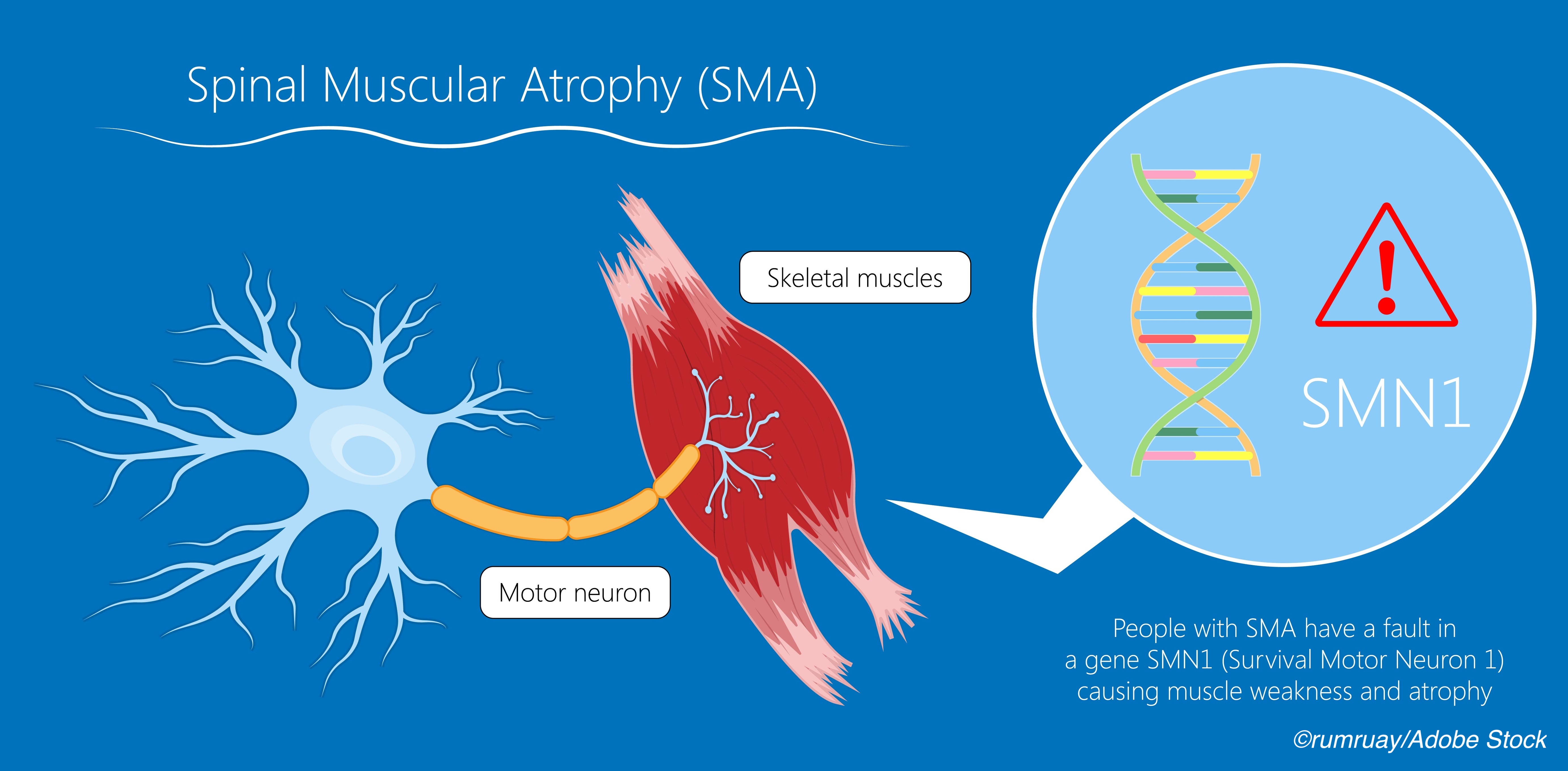
SMA is caused by deletions or mutations in the survival of motor neuron 1 gene (SMN1), resulting in insufficient levels of SMN protein, which is necessary for motor neuron function. Risdiplam is an mRNA splicing modifier that increases systemic SMN protein concentrations by improving the efficiency of SMN2 gene transcription and has been approved by the FDA for treating infants with SMA who are two months of age or older.
In their study, which was published in the New England Journal of Medicine, Baranello and colleagues reported on the results from part 1, which was a dose-finding study.
The primary outcomes of the study were safety, blood SMN protein concentration, dose selection for part 2 of the study, and exploratory outcomes such as the ability to sit without support for at least 5 seconds.
Twenty-one infants were enrolled in the study — 4 in a low-dose group who at month 12 were given a final dose 0.08 mg of risdiplam per kilogram per day, and a high-dose group of 17 infants who were treated with a final dose at month 12 of 0.2 mg per kilogram per day.
At the time of the study’s publication, Baranello and his colleagues found that 7 infants in the high-dose group were able to sit without support for at least 5 seconds, while none of the infants in the low-dose group were able to do so. This was an unexpected finding.
In addition, 9 of the infants in the high-dose group were able to maintain upright head control at all times, and 1 infant in that group was even able to stand. None of the infants in the low-dose group were able to perform these functions. Over a 12-month period none of the infants lost the ability to swallow, and at 12 months, 86% were able to take food orally (exclusively, or in combination with a feeding tube).
The authors also found that 12 infants (52%) achieved scores of 40 or higher on the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND), “a score that is rarely observed in patients with type 1 spinal muscular atrophy.”
However, they added, “it cannot be stated with confidence that there was clinical benefit of the agent because the exploratory end points were analyzed post hoc and could only be qualitatively compared with historical cohorts.”
SMN protein concentrations increased from baseline values of 1.31 ng per milliliter in the low-dose group, and 2.54 ng per milliliter in the high-dose group, to 3.05 ng per milliliter and 5.66 ng per milliliter, respectively, at 12 months.
“In this trial of risdiplam in infants with type 1 spinal muscular atrophy, 24 serious adverse events had been reported as of the clinical data cutoff date,” the study authors noted. “The most common serious adverse events were infections of the respiratory tract, and 4 infants died of respiratory complications; these findings are consistent with the neuromuscular respiratory failure that characterizes spinal muscular atrophy.”
Baranello and colleagues also noted that four weeks after treatment initiation, they observed a median SMN protein level of 2.1 times baseline level in the high-dose cohort. However, there was variation between infants. “The mean exposure to risdiplam at which this increase was observed was the highest mean exposure of risdiplam that did not lead to retinal toxic effects in toxicologic studies in animals,” they wrote. “On the basis of the pharmacokinetic data, the selected dose for part 2 of this study was o.2 mg per kilogram.”
As of this writing, there are three FDA-approved therapies for the treatment of SMA — nusinersin, onasemnogne abeparvovec, and risdiplam. The latter is approved for children with SMA who are age two months or older.
-
Administration of a higher dose of oral risdiplam led to improvements in motor function in some infants with type 1 spinal muscular atrophy.
-
Treatment with risdiplam led to an increased expression of functional SMN protein in infants in both the low-dose and high-dose cohorts of the study.
Michael Bassett, Contributing Writer, BreakingMED™
Baranello reported receiving consulting fees from AveXis, F. Hoffmann-La Roche, PTC Therapeutics, Inc., and Sarepta Therapeutics, Inc., as well as speaker honoraria from PTC Therapeutics.
Cat ID: 130
Topic ID: 82,130,730,130,192,925