The following is a summary of “Approach to the Patient With Congenital Hypothyroidism,” published in the December 2022 issue of Endocrinology & Metabolism by Stoupa, et al.
If identified and treated early, congenital hypothyroidism (CH) is the most prevalent avoidable cause of developmental delay and growth failure in neonates. The thyroid is the first endocrine gland to appear in humans and develop during embryonic life. A first period of embryogenesis and a second phase of folliculogenesis and differentiation, including thyroid hormone synthesis at the later stages, can be used to split thyroid development and maturation into two parts. For a study, researchers sought to outline the therapeutic strategy for treating a kid with CH, emphasizing the diagnostic process and potential difficulties with optimizing thyroid replacement treatment and regenerative medicine.
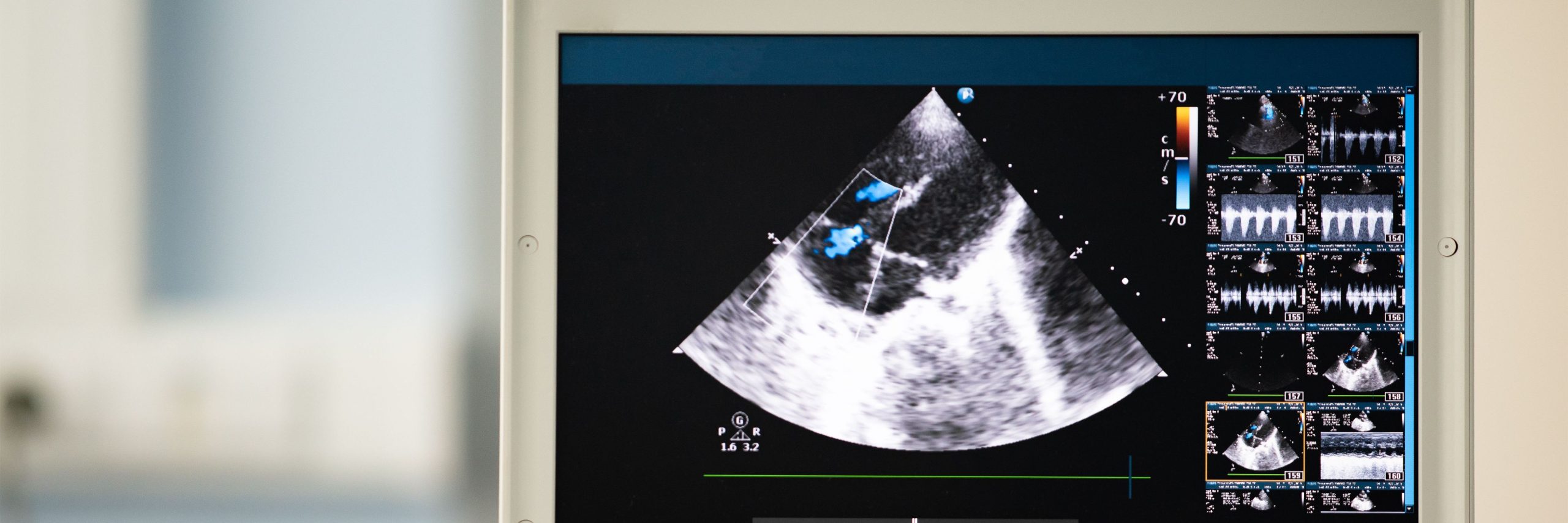
The correct development of the hypothalamic-pituitary-thyroid axis, which takes place during the embryonic and neonatal period, is necessary to regulate thyroid function. Permanent CH results from errors in any of the stages of thyroid development, differentiation, and control. Only one-third of nations had newborn screening programs, which were cost-effective, highly sensitive, and specific in detecting CH. The prevalence of thyroid in situ in primary CH had grown during the past ten years, changing the epidemiology of CH. The formation and function of the thyroid were better understood because of developments in molecular testing. However, only 5% of CH caused by thyroid dysgenesis had a molecular etiology identified.
The review was given from the viewpoint of the case of two daughters who were identified with thyroid ectopy and referred for CH following neonatal screening. New mutations in the TUBB1 gene, linked to big platelets and aberrant platelet physiology, were discovered through genetic testing.
Reference: academic.oup.com/jcem/article-abstract/107/12/3418/6701628